撰稿 | 孙也
责编 | 刘坚 唐倩
2022年7月14日,来自南洋理工大学的Franklin L. Zhong课题组在Science上发表题为”ZAKa-driven ribotoxic stress response activates the human NLRP1 inflammasome”的研究性文章,并提出UVB以及毒素可诱导由ZAKa/MAPK20驱动的核糖毒性应激响应,该响应可进一步促进NLRP1炎症小体的活化。同时建立了作为核糖毒性应激响应(RSR)一个组成部分的炎症小体驱动细胞凋亡新机制。

先天免疫通过保守的传感器蛋白识别病原体相关分子模式(PAMP)和损伤相关分子模式(DAMP)。含NATCH,LRR和PYD结构域蛋白(NLRPs)在细胞内病原体或应激信号的响应下组装炎症体复合体,可导致以caspase-1激活、Gasdermin D(GSDMD)孔形成和IL-1(IL-1)分泌为特征的细胞焦亡。其中,由于不同于啮齿动物的结构域排列,人NLRP1炎症小体传感器引起了人们的广泛注意。蛋白酶抑制剂DPP8和DPP9是唯一已知的可以同时激活啮齿动物和人NLRP1、以及人类炎症小体相关感应蛋白CARD8的分子。
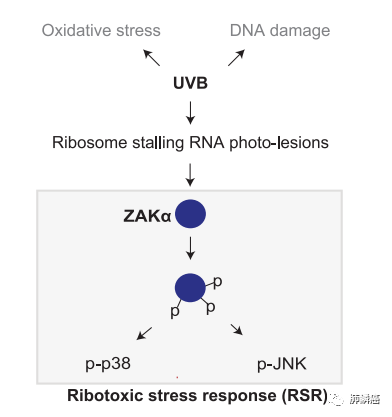
最近的研究表明,人NLRP1可以感知双链病毒RNA、病毒蛋白水解酶和紫外线B(UVB),而这些因素并不会激活啮齿动物的NLRP1s。人NLRP1主要在皮肤和呼吸道上皮细胞中表达,且研究发现NLRP1突变和常见的单核苷酸多态性均与人类皮肤病有关。因此,人NLRP1在皮肤免疫中发挥着独特的作用。波长为280到315纳米的UVB辐射是导致急性晒伤的原因,是迄今为止已确定的所有NLRP1诱因中与皮肤最为密切的。然而,NLRP1感知UVB的分子机制尚不清楚。因此,研究人员通过CRISPR Cas9、免疫印迹等技术揭示了人NLRP1激活的重要机制,包括1)UVB通过损伤RNA诱导NLRP1的活化;2)在UVB活化NLRP1的过程中,RSR激酶ZAKα被激活,并与其下游效应器p38共同磷酸化NLRP1的人类特有的无序连接区NLRP1DR(图一)。
首先,研究人员使用N/TERT-1永生化的人角质形成细胞系(N/TERT),发现UVB照射引起NLRP1依赖的细胞焦亡呈剂量依赖性,如IL-1B分泌、GSDMD裂解、ASC寡聚。用毒性剂量的DNA损伤化学物质如依托泊苷或过氧化氢等处理N/TERT细胞未能诱导NLRP1依赖的IL-1β分泌。因此,DNA损伤或自由基单独造成的氧化损伤都不是UVB诱导的NLRP1炎症体激活的主要驱动因素。因此,研究人员假设UVB驱动的RNA损伤激活了NLRP1炎症体。团队用核苷类似物4-硫代尿苷(4-SU)对细胞进行预处理,其可以选择性地使RNA对紫外线A(UVA)辐射敏感,而UVA不会对未经修饰的核酸造成损害。研究人员发现只有在经4-SU处理的N/TERT细胞中,UVA才能引起IL-1β的分泌。这也证明了RNA的光损伤可能是紫外线诱导角质形成细胞NLRP1激活的上游信号(图二)。
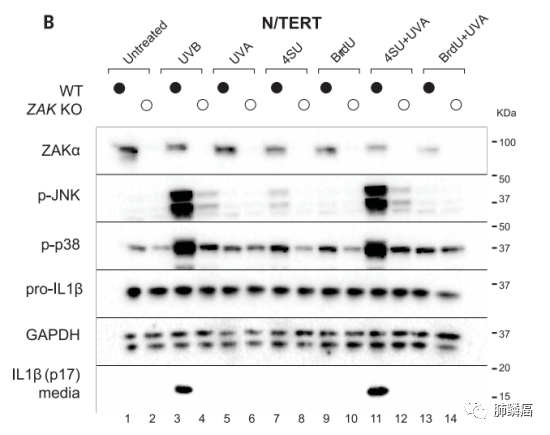
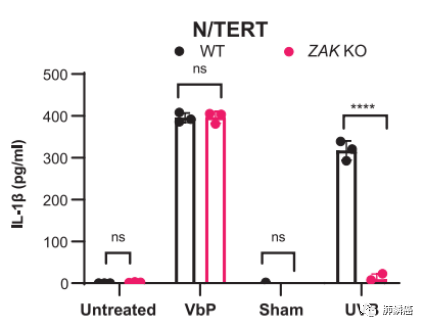
最近的研究发现UVB通过ZAKα激酶触发应激活化蛋白激酶(SAPK)激活。ZAKα激酶可检测由于UVB诱导而停滞的核糖体并被激活,进而磷酸化下游的SAPKs。这一途径被称为核糖毒性应激响应(RSR)。UVB诱导N/TERT角质形成细胞的RSR激活,体现为早期的ZAKα、P38和JNK的磷酸化,以及后期的ZAKα降解。然而在CRISPR/Cas9 ZAK基因敲除(KO)的N/TERT细胞中,p38和JNK的磷酸化则完全被取消,且ZAKα的抑制会阻碍IL-β的分泌。这体现了ZAKα的核糖体结合和RSR感应结合域是RSR和NLRP1炎症体激活所必需的(图三)。并且,课题组使用ZAKα的激活毒素如茴香素(ANS)等观察到ZAKα的磷酸化且促进分泌IL-1β和GSDMD。说明ANS等激活毒素可以起到激活NLRP1炎症小体的作用,但并非通过抑制翻译作用。团队发现,敲除ZAKα、NLRP1可消除N/TERT细胞中ANS依赖的细胞焦亡。ANS还可以在包皮角质形成细胞、支气管上皮细胞和主动脉内皮细胞中诱导炎性小体驱动的细胞凋亡。然而,在MV-4-11细胞中,ANS并未发挥作用,因为该细胞中CARD8作为主要的炎症小体传感器。因此,激活ZAKα的核毒素,如ANS,起到了真正的NLRP1激活剂的作用。
接下来,研究人员分别用人NLRP1、NLRP1敲除PYD、鼠NLRP1B(muNLRP1B),或人CARD8重组了NLRP1 KO N/TERT细胞,并发现ANS不能以muNLRP1依赖的方式、仅能在人NLRP1过表达或NLRP1敲除PYD的细胞中诱导IL-1β。人NLRP1含有一个独特的N-末端延长结构域,称为NLRP1DR。该结构域不存在于鼠NLRP1和CARD8结构中。课题组发现,NLRP1DR的缺失,而不是NLRP1 PYD的缺失,消除了UVB/ANS触发的细胞焦亡。这说明NLRP1DR是ZAKα依赖的NLRP1炎症体激活所必需的(图四)。
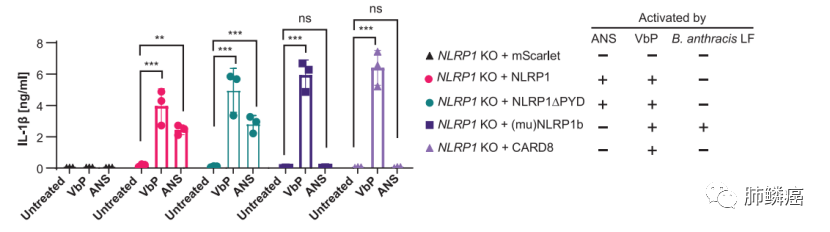
图四:重组NLRP1 KO N/TERT细胞和CARD8细胞经ANS处理后的IL-1β分泌情况
NLRP1DR是一种GFP融合蛋白(NLRP1DR-GFP),每当UVB或ANS处理细胞时,研究人员通过免疫印迹观察到NLRP1DR-GFP的明显条带移动,这被证实是由磷酸化的NLRP1DR引起的。在所有被测试的药物中,只有激活ZAKα的化合物能诱导这种NLRP1DR过度磷酸化。因此,当被过度表达或核糖毒性应激响应时,ZAKα会过度磷酸化NLRP1DR中的NLRP1。且通过突变NLRP1DR片段中的丝氨酸/苏氨酸残基发现,NLRP1DR中单个磷酸化位点突变即可控制着ZAKα驱动的NLRP1激活(图五)。
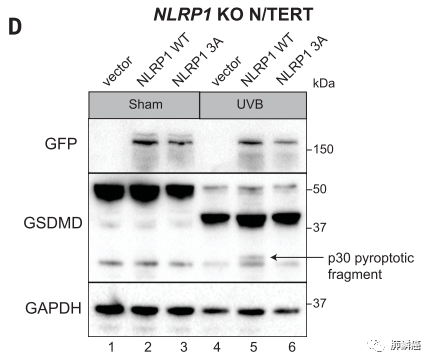
图五:UVB照射后表达全长NLRP1 T178A、S179A或T180A(3A)突变体的NLRP1-KO N/TERT细胞的GFP和GSDMD免疫印迹
由于ZAKα激活了多个SAPKs,课题组测试了其他激酶是否也参与了ZAKα诱导的NLRP1激活。团队发现多个p38抑制剂阻断ANS和UVB诱导的IL-1β的分泌。然而,P38抑制剂对ANS诱导的NLRP1DR过度磷酸化只有轻微的影响,且会被ZAKα抑制剂所废除。因此,这也表明P38激酶参与了RSR诱导剂诱导的NLRP1炎症小体的激活,但作用不如ZAKα那么关键。由于p38α和p38β在RSR信号通路中位于ZAKα下游,因此研究人员假设ZAKα是使NLRP1磷酸化的起始激酶,而p38激酶随后加强或放大这一反应。
2021年1月8日,来自南洋理工大学生物科学学院的 Bin Wu课题组在Nature Communications上发表题为Structural basis for distinct inflammasome complex assembly by human NLRP1 and CARD8的研究性文章,揭示了NLRP1-CARD and CARD8-CARD的冷冻电镜结构,该结构使 NLRP1 和 CARD8 能够区分 ASC 和促半胱天冬酶-1,为人类NLRP1和CARD8的激活机制提供了结构见解。
简评
UVB通过诱导细胞RNA光损伤使核糖体停滞,进而激活RSR激酶ZAKα,ZAKα再与其下游效应器p38共同磷酸化NLRP1。这为人类皮肤角质形成细胞对UVB的感知的理解提供了新角度。同时,人NLRP1是一种多功能的传感器蛋白,可以通过其离散的结构域整合多个信号。本文也扩大了已知的人类NLRP1激动剂的保留范围,包括多种微生物核毒素,如ANS。
---Ye Sun
原文链接:
doi: 10.1126/science.abl6324
参考文献:
2.Gong, Q., et al. Structural basis for distinct inflammasome complex assembly by human NLRP1 and CARD8. Nat Commun. Jan 8, 2021.




