 55岁男性,运动障碍。本文译自《Neurology》杂志第97卷第23期,发表于2021年12月7日。点击下载文献原文
55岁男性,运动障碍。本文译自《Neurology》杂志第97卷第23期,发表于2021年12月7日。点击下载文献原文
Section 1
病例介绍:
55岁男性,既往焦虑、抑郁、精神分裂症、PTSD病史,长期口服喹硫平及曲唑酮治疗。40岁时曾诊断为抽动秽语综合症。长期肝酶(AST、ALT)升高数十年。患者近期从精神病院出院后在康复医院就诊,期间出现怪诞行为。如在地板上吃东西、向工作人员吐痰、伴吞咽困难、反流等。消化内科评估后未发现胃肠道器质性病变。神经科医师会诊考虑多动症,后患者转我院治疗。入院初步印象:消瘦,多动,以躯干及双上肢为主,坐时明显,患者无法抑制上述动作。伸舌坚持5秒不能完成。眼动、共济、感觉等正常。可独自行走,但类似于蹒跚步态。患者否认家族史。入院AST304U/L,ALT154U/L。
Q:患者是哪种类型的运动障碍?可能的鉴别诊断?
译者注:看表现像舞蹈症,要有视频就更好了。症状上看除了运动过多还有新发精神行为异常如吐痰、地板上吃东西等,加上都是新发症状,病因考虑感染、免疫、药物相关等。
Section 2
患者症状符合舞蹈症,即肢体不规则、无节律和无目的的不自主运动,上肢重于下肢,远端重于近端。舞蹈症的鉴别诊断包括影响基底节或丘脑底核的结构性疾病(肿瘤、脱髓鞘、血管病)、中毒代谢病(非酮性高血糖、电解质紊乱、CO中毒)、药物(抗精神病药物迟发性运动障碍)、自身免疫病(SLE、抗磷脂综合症)、遗传(亨廷顿病、神经棘红细胞增多症、肝豆状核变性)、感染(弓形虫病、HIV脑病、CJD)、副肿瘤综合征(抗CRMP-5脑炎、抗NMDA脑炎等)其他原因(真性红细胞增多症、心因性疾病)等。
该患者舞蹈症属于全身性,非偏侧性,可暂除外结构性病变如血管病、高血糖等。该患者发病隐匿(初看第一段病史像急性/亚急性起病,但真正遇到舞蹈症的病人通过病史判断急性/恶急性/慢性/隐匿性起病是不难的,译者注),考虑遗传因素可能性大。
Q:下一步检查?
译者注:先排遗传性的病因,查头MR、血常规、血涂片、K-F环、铜蓝蛋白等。
Section 3
患者精神行为异常、运动障碍、肝酶升高,高度怀疑肝豆状核变性(威尔逊病WD)。肝豆常见的运动症状是震颤和共济失调。该患者血清铜及铜蓝蛋白检测正常。完善头MR,发现双侧尾状核萎缩。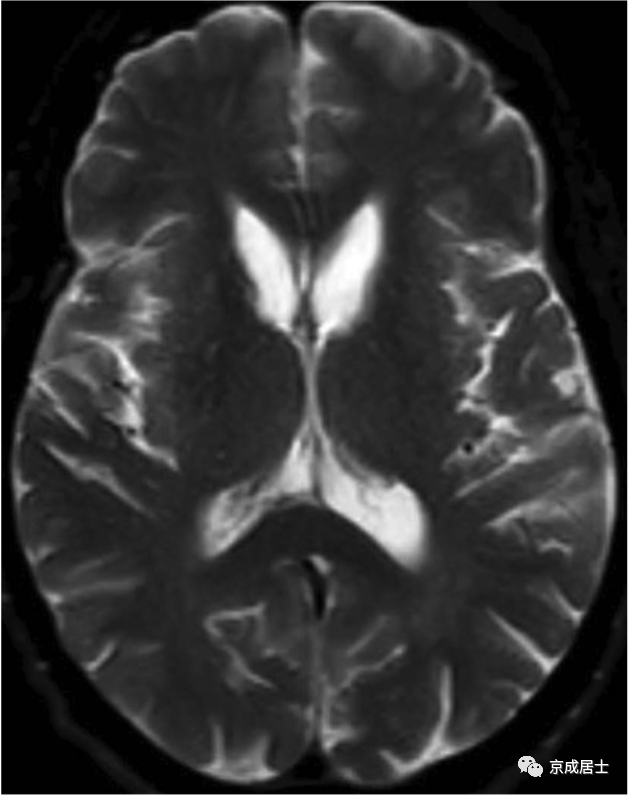
Q:下一步?
译者注:不像肝豆,再查下K-F环,如果也没有,肝豆的基因也没必要查了。应考虑神经棘红细胞增多症的可能。
Section 4
结合头MR双侧尾状核头萎缩的情况,考虑亨廷顿病和神经棘红细胞增多症的可能。亨廷顿病基因检测CAG重复序列正常,血涂片示20%棘细胞,最终诊断神经棘红细胞增多症明确。
讨论
神经棘红细胞增多症临床特征有舞蹈症、肌张力障碍、唇咬伤、抽搐、精神行为异常,血涂片可见棘红细胞,平均35岁发病,可伴CK升高。值得注意的是,除了神经棘红细胞增多症,有些退行性运动障碍病也可出现棘红细胞增多,如泛酸激酶相关神经变性PKAN和亨廷顿病2型HDL2。PKAN是常染色体隐性遗传病,多儿童期发病,头MRI可见双侧苍白球高信号,呈“虎眼征”。HDL2则成年期发病,常见于非洲裔人群。
译者注:笔者之前管过1例神经棘红细胞增多症的病例,印象最深刻的就是在唇部不自主运动和舌咬伤。有关神经棘红细胞增多症的相关情况可以参考这篇链接(20190911神经肌肉遗传科教学查房—VPS13A基因单杂合突变导致常染色体显性遗传舞蹈症-棘红细胞增多症)。这个病例对笔者最大的启示是遗传性舞蹈症的鉴别诊断。
版权声明:文章来源见出处,版权归原作者,转载仅用于学术交流学习,如有侵权,请联系我,我将第一时间删除并致歉。




